
The Kaderali Lab
Institute of Bioinformatics · University Medicine Greifswald · University of Greifswald
Menu:
Bioinformatics and Statistical Data Analysis
Modern experimental high-throughput platforms generate large datasets, for which sophisticated analysis tools are required. We develop computational and statistical methods to normalize and analyze complex data, from basic statistical processing to the downstream bioinformatics analysis. We work with a wide range of different data types, stemming, for example, from Next Generation Sequencing, genome wide image based RNAi Screening, Gene Expression Microarrays, aCGH, Mass Spectrometry, Flow Cytometry, Life Cell Imaging, Yeast-2-Hybrid screening, Western and Northern Blotting, and other platforms.
Projects are typically carried out in close collaboration with experimental groups, and methods developed are directly applied to solve relevant biological problems. We furthermore offer Statistical consulting as a service to members of the Medical Faculty of Greifswald University and to collaboration partners.
High Throughput Sequencing

Next generation sequencing platforms enable the deciphering of full genomes, of genome wide expression, genomic aberration or methylation patterns in a cost-efficient, high-throughput manner. Data coming from NGS platforms require special algorithms for the alignment and/or mapping of the short sequence reads, and sophisticated tools are needed for the downstream bioinformatics analysis of the vast amounts of data generated in large scale sequencing projects.
In close collaboration with biological groups and sequencing facilities, we develop analysis methods for sequencing projects, and apply them to data from collaboration partners. Questions addressed in our group include, for example, the identification of genomic aberrations in cancer, of point mutations underlying developmental disorders, species identification from their mitochondrial DNA, and identification of ageing-associated methylation changes.
Microbiome Data Analysis
Using 16S RNA or metagenome sequencing, we can characterize microbial composition in different body nitches such as in our gut, in our mouth or nose, airways, lung, and on our skin. These data can then be used, for example, to correlate microbiome composition with disease risk, and to characterize novel mechanisms of disease development and progression.
We develop computational tools to analyze both 16S RNA as well as shotgun metagenome sequencing data, and apply these tools in collaboration with experimental and clinical groups to study health and disease.
OMICs Data Analysis

OMICs data have revolutionized biology in the last decade. OMICs data have been shown to correlate with therapy response and patient survival in many disease and can assist a clinician in evaluating treatment options, and genes found to correlate with survival may hint at novel targets for drug design.
Whether Gene Expression Microarrays, RNA or DNA sequencing, ChIP-on-Chip, ChiP-Seq, Metabolomics, Mass Spectrometry or other platforms, analysis of these data is still an art and requires sophisticated normalization and analysis tools for robust identification of differentially expressed genes, proteins and pathways. Due to the high dimensional nature of the experimental data, computational tools are needed for data analysis. We develop and apply such methods in close collaboration with our experimental partners.
Multi-OMICs data integration
Integration of data stemming from different experimental platforms is becoming increasingly difficulty. Vast amounts of data are being generated, but interpreting and analyzing this data and integrating it with data from others studies and other platforms is becoming more and more problematic.
We develop and apply bioinformatics tools for multi-OMICs data integration, for example by integrating OMICs data with protein interaction and pathway information, thus mapping available experimental information to the underlying biological processes. Sophisticated computational algorithms are required for this task, to maximally extract information from experiments.
Functional information gain through sequence analyses
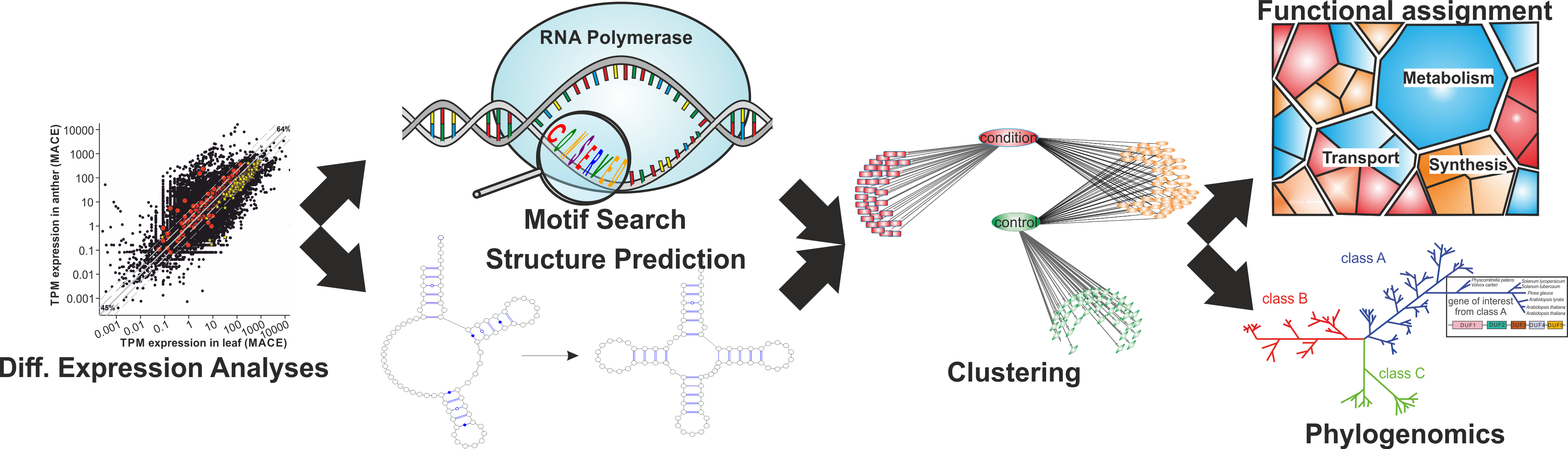
Data mining of sequence information based on different -Omics sources allows the identification of expressed ncRNAs as well as proteins and isoforms. In relation to the adaptation to abiotic and biotic stresses or developmental effects, these post-transcriptional changes, alternative splice forms and ncRNAs can have massive effects on regulatory mechanisms and pathways, which can be of unknown function so far.
To get insight in the function of these expressed RNAs or proteins structure prediction, motif search and functional domain architecture are important. By this, conservation of specific features can be used for classification via Machine learning algorithms to draw conclusions about the regulatory mechanisms of single marker as well as specific groups of sequences sharing the same features. Furthermore, clustering due to phenotypic features or common motifs and phylogenomics analysis can help to understand the evolutionary context and changes due to environmental conditions.